Jaundiced baby- what should I do as a pediatrician?
Review
Review
![]()
Abstract:
Neonatal cholestasis is an emergency situation. Many of these cases are still diagnosed late and inadequately treated. Biliary atresia is most common condition which presents as neonatal cholestasis. Prompt detection of a case, a guided investigational approach, specific management and timely referral is very crucial. A concise review is presented for a pediatrician to guide about the appropriate measures to be taken to offer the best possible outcome for a neonate with cholestasis.
Key words:
Neonatal cholestasis, biliary atresia, approach
Abbreviations:
INH- Idiopathic neonatal hepatitis, GGTgamma- glutaryl transferase, INR- internationally normalized ratio, CMV- cytomegalovirus, PFICprogressive intrahepatic familial cholestasis.
Inroduction: Jaundice is a common problem in newborn affecting almost 25-30% of near term babies. However, majority of them have transient unconjugated hyperbilirubinemia due immaturity of hepatic enzyme glucuronyltransferase. A small portion of them had cholestasis. Neonatal cholestasis means decrease in bile flow and elevation of conjugated bilirubin. Conjugated hyperbilirubinemia is generally defined as a conjugated or direct bilirubin level greater than 1 mg/dL when the total bilirubin is less than 5 mg/dL or more than 20% of the total bilirubin if the total bilirubin is greater than 5 mg/dL.1 Conjugated hyperbilirubinemia is never physiologic or normal. Cholestatic jaundice is seen in 1 in 2500 in western countries and 26% of all chronic liver disease in children from India.2 The process occurs as a result of impaired bile formation by the hepatocytes or from obstruction to the flow of bile through the intrahepatic and/ or extrahepatic biliary tree. In the neonate, the clinical and laboratory features of the many liver diseases presenting with cholestasis are quite similar. An important focus in approaching neonatal cholestasis is a prompt differentiation between intrahepatic and extrahepatic cholestasis and, if possible, to establish a specific diagnosis. Most of the neonates with biliary atresia appear well except jaundice. This lures parents and physician to delay the investigations. Extrahepatic cholestasis like biliary atresia is amenable to surgical correction and early surgery (within 8 weeks) is extremely important for successful bile drainage and prolonged survival. Mieli-Virgani et al demonstrated that more than 80% babies with BA became jaundice free when the portoenterostomy was done before 60 days of age, while the success rate of establishing biliary drainage dropped to 25-35% only when the surgery was done after 60 days of age.3
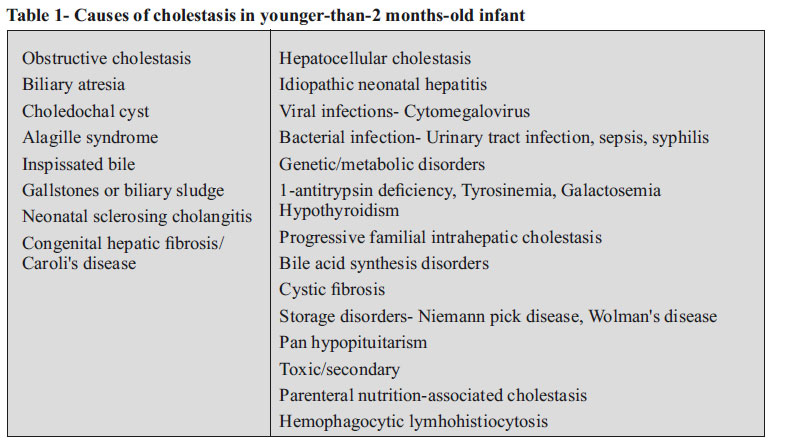
No screening test can predict which infant will develop cholestasis. So, detection of cholestasis depends on clinical recognition of yellow colored eyes, diaper staining (dark colored urine) and/ or pale stools by parents. Causes of cholestasis in younger infants (< 2 months of age) are mentioned in table 1. A cost-effective, quick, simplified and appropriate management protocol of neonatal cholestasis in developing countries was published in Indian Pediatrics.4 The number of distinct diseases presenting as cholestasis is greater in neonates than at any other time of life. So-called Idiopathic neonatal hepatitis (INH) was the most common diagnosis. But in recent era with new diagnostic modalities available, the diagnosis of INH is decreasing and rarer diseases are becoming more common. There is no alternative to a thorough clinical history and physical examination, which can give many diagnostic clues. Observing the stool color of each baby is very important. All babies while being discharged after birth should have a documentation of their stool color in the discharge card. In Taiwan, a national screening program has been implemented through which an infant stool color card is placed into the child health booklet given to every neonate. This program has increased the national rate of the Kasai operation performed before 60 days of age from 49% to 66%, and it has increased the 3-month jaundice-free rate after the Kasai operation from 35% to 61% (p < 0.001). In addition, the 5-year jaundice-free survival rate with native liver increased from 27% to 64% (p < 0.001) and the 5-year overall survival rate increased from 56% to 89% (p < 0.001).5 Such a national effort is grossly lacking in India. A child with suspected neonatal cholestasis should first have a liver function test including gamma-glutaryl transferase (GGT) and internationally normalized ration (INR) (after vitamin K). A presence of persistent clay colored stool has 79% accuracy of diagnosing biliary atresia.6 After confirmation of conjugated hyperbilirubinemia, synthetic function (normal or delayed INR), clinical status (as sick or stable); the baby should be evaluated as per the focused and rapid investigational approach (Figure 1).4 Ultrasonographic evaluation of liver should be specifically looked for gall bladder contractility, triangular cord signs, dilatation of biliary system, splenic malformations, inspissated bile, any features of cirrhosis, collaterals and ascites. Liver biopsy plays a very crucial role in diagnosing biliary atresia with sensitivity and specificity of 88.2 each.7 A nuclear scan is useful to exclude biliary atresia rather than diagnosing it. Features of biliary atresia which are found on liver biopsy are portal tract widening, portal tract fibrosis, bile ductular plugs and bile ductular proliferation.7 Apart from biliary atresia, many other diseases like progressive familial cholestasis, Alagille syndrome, congenital hepatic fibrosis, Niemannpick disease, cytomegalovirus (CMV) hepatitis, Alpha-1 antitrypsin deficiency. A clue to a possible metabolic disease can be picked up by finding micro vesicular hepatic steatosis. Any sepsis should be treated promptly and etiological work-up should be initiated. In a baby with neonatal cholestasis with cirrhosis, always suspect biliary atresia, Galactosemia, Tyrosinemia, progressive intrahepatic familial cholestasis (PFIC)-2, bile acid synthetic disorders. For a neonatal cholestasis with deranged INR, one should think of metabolic disorders like Galactosemia, Tyrosinemia, and neonatal hemochromatosis. In a baby suffering for neonatal cholestasis with ascites, one should look for Galactosemia, Wolman’s disease, Tyrosinemia, spontaneous perforation of bile ducts. The etiology wise work up is listed in table 2.
Even if the specific diagnosis or treatment is not possible, infants with progressive liver disease usually benefit from optimal nutritional support and medical management of complications of cholestasis and possibly cirrhosis until liver transplantation is performed. Sufficient calories and protein intake (Calories- 125 % of the RDA and protein 2-3 g/kg/ day) along with medium chain triglycerides (MCT) oil should be given. The optimum nutritional management is summarized in table 3.8 Specific management of individual condition is summarized in table 4. It’s worthwhile to note that many of these are treatable.
To summarize, neonatal cholestasis is an emergency condition and prompt investigation and referral is must. A suspected case of biliary atresia must be picked up and diagnosed before 4-6 weeks of age and should be offered Kasai portoenterostomy. Apart from biliary atresia, many of the conditions are treatable and warrants complete investigation.
References:
1) Moyer V, Freese DK, Whitington PF, et al. Guideline for the evaluation of cholestatic jaundice in infants: recommendations of the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr 2004; 39: 115- 128.
2) Yaccha SK, Sharma BC, Khanduri A, Srivastava A. Current spectrum of hepatobiliary diseases in Northen India. Indian Pediatr 1997; 34: 885-90.
3) Mieli-Vergani G, Howard ER, Portman B, Mowat AP. Late referral for biliary atresia – missed opportunities for effective surgery. Lancet 1989; 1: 421-3.
4) Consensus report on neonatal cholestasis syndrome.Pediatric Gastroenterology Subspecialty Chapter of Indian Academy of Pediatrics. Indian Pediatr 2000; 37: 845-51.
5) Lien TH, Chang MH, Wu JF, et al. Effects of the infant stool color card screening program on 5-year outcome of biliary atresia in Taiwan. Hepatology 2011; 53: 202-208.
6) Poddar U, Thapa BR, Das A, Bhattacharya A, Rao KL, Singh K. Neonatal cholestasis: differentiation of biliary atresia from neonatal hepatitis in a developing country. Acta Paediatr 2009; 98(8):1260-4.
7) Rastogi A, Krishnani N, Yachha SK, Khanna V, Poddar U, Lal R. Histopathological features and accuracy for diagnosing biliary atresia by prelaparotomy liver biopsy in developing countries. J Gastroenterol Hepatol 2009; 24: 97-102.
8) Bhatia V, Bavdekar A, Mathai J, Waikar Y, Sibal A. Management of Neonatal Cholestasis: Consensus Statement of the Pediatric Gastroenterology Chapter of Indian Academy of Pediatrics. Indian Pediatr 2014; 51: 203-10.




Issue: July-September 2015 [Volume 4.3]