A Rare Case : Sjogren-Larsson Syndrome
Case Report
Case Report
![]()
Abstract:
A 2.5 year boy was brought with spastic diplegia, delayed attainment of motor developmental milestones and congenital ichthyosis. He had borderline intelligence. Magnetic resonance imaging showed bilateral hyperintensities on T2WI and FLAIR sequences with MR spectroscopy showing a characteristic lipid peak at 1.3ppm. clinical and MR findings resemble those of Sjogren-larsson syndrome.
Keywords: Sjogren-Larsson Syndrome, Congenital Ichthyosis, Spastic Diplegia
Introduction:
Sjögren-Larsson syndrome is an autosomal recessive inborn error of metabolism due to an abnormality of fatty alcohol oxidation that results from a deficiency of fatty aldehyde dehydrogenase (FALDH3A2).1
The clinical picture of Sjögren-Larsson syndrome consists of ichthyosis, cognitive impairment, and spasticity. The ichthyosis is generalized but is accentuated on the flexures and the lower abdomen and consists of erythroderma, fine scaling, larger plate-like scales, and dark hyperkeratosis. Most individuals have palmoplantar hyperkeratosis. Glistening dots in the foveal area are a cardinal ophthalmologic sign. About half the patients have primary retinal degeneration. Motor and speech developmental delays are usually noted before 1 year of age, and spastic diplegia or tetraplegia, epilepsy, and intellectual disability generally become evident in the first 3 years of life. Elevation of urinary leukotriene B4 (LTB4) may provide an easier approach to diagnosis.1
MR imaging shows defective myelination and a mild persistent myelin deficit. A zone of increased signal intensity is seen in the periventricular white matter on T2-weighted images. MR spectroscopy of white matter may reveal a characteristic sharp, narrow peak of lipid at 1.3 ppm. MR imaging and proton MR spectroscopy of gray matter may be normal.2
We are reporting a case of Sjogren-larsson syndrome to discuss its clinical and radiological features.
Case Report:
A 2 and half year boy, born preterm at 8 months of gestation, from a non-consanguineous marriage, was brought with the complaints of scaly pruritic skin lesions over upper and lower limbs since birth and inability to walk. He has history of episodes of afebrile seizures. He had mild developmental delay with developmental quotient (DQ) is 70.
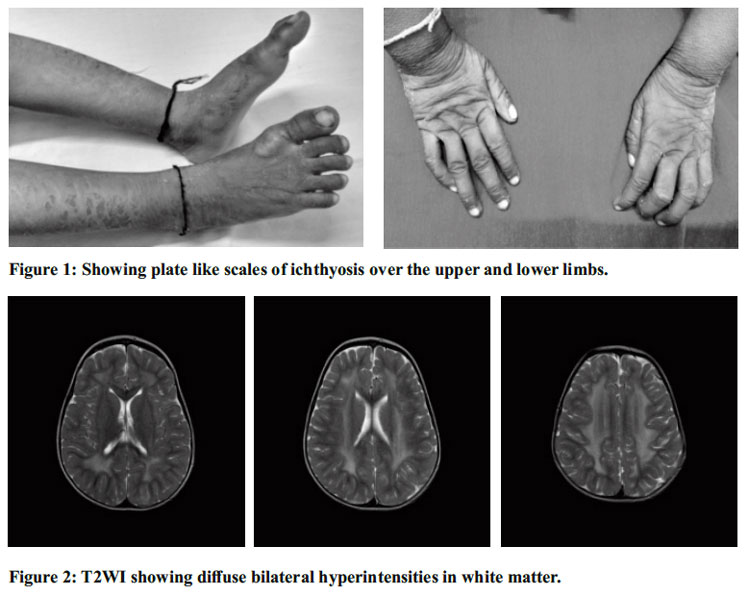
On examination, the child was conscious, playful, and dry, plate-like scales were present over the dorsum of hands, around the neck, axilla, abdomen, and dorsum of feet with relative sparing of the face suggestive of ichthyosis. Neurocutaneous markers were absent. Contractures were present at the knees. Central nervous system examination showed the child to be alert and oriented to person. Cranial nerve examination was normal. Hypertonia was more in lower limbs than upper limbs with exaggerated deep tendon reflexes. Babinski reflex was positive. Power was grade 4 in upper limbs and grade 3 in lower limbs. Involuntary movements were absent. No signs of meningeal irritation or cerebellar involvement. Cardiovascular system, respiratory system and abdominal system examination were unremarkable. The clinical triad of Congenital ichthyosis, Spastic diplegia and Mental retardation aroused the suspicion of Sjogren-Larsson syndrome.
Complete blood picture, liver function test and renal function tests were within normal limits.
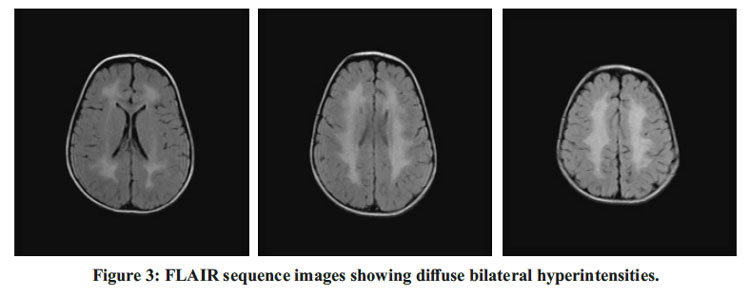
Fundoscopy was normal. MRI done showed diffuse bilaterally symmetrical periventricular hyperintensities on T2WI and FLAIR with no evidence of diffusion restriction and sparing of subcortical Ufibres. MR Spectroscopy showed a prominent lipid peak at 1.3 ppm. EEG done showed evidence of intermittent bursts of spike wave discharges lasting for 2-3 seconds subictogenic activity. BERA done showed no hearing impairment. Histopathology of skin revealed epidermal hyperkeratosis showing peaks and valley appearance, hypertrophied squamous lining with loss of rete pegs, basal layer showing mild vacuolation and dermis showing sparse inflammation and mild edema.

Discussion:
Sjögren-Larsson syndrome is a rare autosomal recessive inborn error of metabolism. Sjogren-Larsson syndrome was originally described in Sweden.2 In 1957, Sjogren and Larsson described a series of patients who suffered from a severe disorder with three cardinal features: congenital ichthyosis, mental retardation and spastic diplegia or tetraplegia.2 The worldwide prevalence is <0.1 per 100,000.3
The disease is caused by mutations in the ALDH3A2 gene on chromosome 17p11.2 that encodes fatty aldehyde dehydrogenase (FALDH), an enzyme that catalyzes the oxidation of longchain aldehydes derived from lipid metabolism. The potential disease mechanisms leading to symptoms include: accumulation of toxic fatty aldehydes that form covalent adducts with lipids and membrane proteins; physical disruption of multi-lamellar membranes in skin and brain; abnormal activation of the JNK cell signaling pathway; and defective farnesol metabolism resulting in abnormal PPAR-? dependent gene expression.4
The clinical triad of this syndrome has congenital ichthyosis, spastic diplegia or tetraplegia and mental retardation. Most common differential diagnosis of congenital ichthyosis include Harlequin ichthyosis, Trichothiodystrophy, Neutral lipid storage disease, Conradi-Hünermann-Happle syndrome, Gaucher disease, Keratitis-Ichthyosis- Deafness syndrome and Netherton syndrome and X-linked ichthyosis. Our patient’s skin lesions were concentrated in the neck, flexures and lower abdomen, in which regions the scales were often dark.5 The non-scaly hyperkeratosis produced characteristic and easily visible skin markings.5 The cutaneous manifestations are the result of abnormal lipid accumulation in stratum corneum and granular cells.5,6 Our patient manifested with spastic diplegia and intellectual disability. Our patient’s other CNS manifestations include delayed speech, epilepsy, and intellectual disability. Ophthalmology examination was normal, although, retinal abnormalities are common but usually are seen after 3 years of age which include perifoveal glistening crystalline deposits in ganglion cell layer and inner plexiform layer, microcysts in fovea, and markedly reduced macular pigment.7 These changes, secondary to Mueller cell degeneration and deficiency of retinal carotenoids, are associated with impaired vision and photophobia.8
Neuroimaging has certain prominent features in this syndrome – demyelination and leukoence phalopathy. Willemsen MA et al.,9 observed that patients have a zone of abnormally high signal intensity in the periventricular white matter on T2 weighted images extending from the frontal aspect to the occipital aspect and may have either a frontal or, more often, a parieto-occipital predominance which was seen in our patient’s MRI as shown in figure 2. This zone may have a mildly decreased or normal signal intensity on T1-weighted images.9 Corpus callosum is involved in some patients. The subcortical white matter is unaffected, and the spared zone is distinct from the unmyelinated U fibers. The brainstem and cerebellum are always spared.9 The hyperintense periventricular white matter exhibits a prominent, narrow lipid peak at 1.3ppm representing an abnormal accumulation of lipids due to FALDH deficiency on MR Spectros copy. 9 The diagnostic suspicion is confirmed by targeted sequencing of the ALDH3A2 gene. The biochemical defect can be demonstrated by direct estimation of FALDH activity in leucocytes and cultured skin fibroblasts.
The management of the affected individuals involves multidisciplinary team of neurologists, ophthalmologists, dermatologists, and rehabilitation specialists, among others. Dietary modification to reduce total fat intake and to increase the linoleic/linolenic acid ratio has limited benefits. Symptomatic treatment of ichthyosis consists of applying emollients, keratolytics, and topical calcipotriol.10
Any child with congenital icthyosis, spastic diplegia/tetraplegia and mental retardation should be evaluated for Sjogren-larsson syndrome.
References:
1. Nelsons textbook of paediatrics, 20th edition. 2015; p3172-3173.
2. Willemsen MA, IJlst L, Steijlen PM, et al. Clinical, biochemical and molecular genetic characteristics of 19 patients with the Sjogren- Larsson syndrome. Brain 2001; 124:1426- 1437.
3. Raghuveer TS, Garg U, Graf WD. Inborn errors of metabolism in infancy and early childhood: An update. Am Fam Physician. 2006;73:1981-90.
4. Rizzo, William B. “Genetics and Prospective Therapeutic Targets for Sjögren-Larsson Syndrome.” Expert Opinion on Orphan Drugs 4, no. 4 (April 2016): 395-406.
5. Jagell, S., and S. Lidén. “Ichthyosis in the Sjögren-Larsson Syndrome.” Clinical Genetics 21, no. 4 (April 1982): 243-52.
6. Rizzo WB. Fatty aldehyde and fatty alcohol metabolism: Review and importance for epidermal structure and function. Biochim Biophys Acta. 2014;1841:377-89.
7. Fuijkschot J, Cruysberg JR, Willemsen MA, Keunen JE, Theelen T. Subclinical changes in the juvenile crystalline macular dystrophy in Sjogren-Larsson syndrome detected by optical coherence tomography. Ophthal mology 2008; 115:870-875.
8. Fuijkschot J, Theelen T, Seyger MM, et al. Sjogren Larsson syndrome in clinical practice. J Inherit Metab Dis 2012;35:955-962.
9. Willemsen MA, Van Der Graaf M, Van Der Knaap MS, et al. MR imaging and proton MR spectroscopic studies in Sjogren-Larsson syndrome: characterization of the leukoencep halopathy. Am J Neuroradiol 2004;25: 649- 657.
10. Nagappa, Madhu, Parayil S. Bindu, Shwetha Chiplunkar, Neelesh Gupta, Sanjib Sinha, Pavagada S. Mathuranath, Rose D. Bharath, and Arun B. Taly. “Child Neurology: Sjögren- Larsson Syndrome.” Neurology 88, no. 1 (January 3, 2017): e1-4.
Issue: July-September 2017 [Volume 6.3]