Rubinstein-Taybi Syndrome
CASE REPORT
Corresponding author: Dr.Jayant Vagha ,Professor and Head of Department Department of Pediatrics, Jawaharlal Nehru Medical College and Acharya Vinoba Bhave Rural Hospital, Sawangi (Meghe), Wardha.EMAIL:
Received: 23 th August,2018; Reviewed:24th October 2018, Accepted:21 th December 2018
Citation Of Article: Jayant Vagha ,Ashish Verma, Abhishek Kalwani , Ramnath Reddy, Anjali Kher; Rubinstein-Taybi Syndrome; New Indian Journal Of Pediatrics 7:4
CASE REPORT
![]()
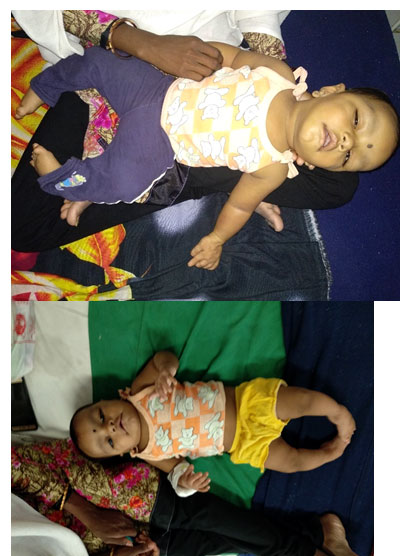
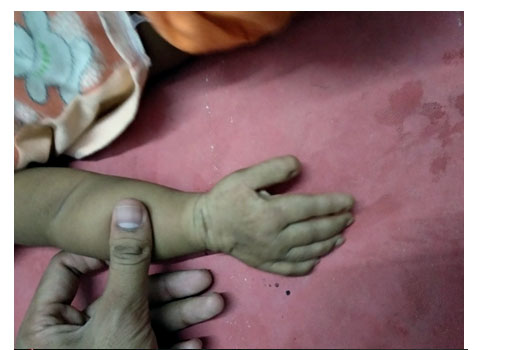
ABSTRACT: Rubinstein-Taybi Syndrome is a rare genetic disorder with characteristic featuresincluding downward slanting palpebral fissures, broad thumbs and halluces,and mental retardation. Systemic features may involve cardiac, auditory,ophthalmic, endocrine, nervous, renal and respiratory systems. This syndromeis sporadic in nature and has been linked to microdeletion at 16p 13.3 encoding CREB-binding protein gene (CREBBP). We report a 6-month-old male, who has congenital talipus equino varus, with downward slanting palpebral fissures towardthe ears, hypertelorism, short stature, beaked nose, micrognathia, large toes and broad thumbs.
Keywords: Rubinstein-Taybi Syndrome (RSTS), Broad thumb and halluces, CTEV.
Introduction: Rubinstein- Taybi Syndrome (RSTS) was initially reported by Michael et al. in1957 as the broad thumb-hallux syndrome and then was described by Rubinstein and Taybi in 19631 in children with broad thumbs and toes, facial abnormalities and short stature. Since then, there have been over 250 cases documented in the literature. It has been estimated that 1 per 300-500 institutionalized persons with mental retardation over age 5 have this syndrome. Male and female individuals are affected at equal rates2.Typical facial features include downward slanting palpebral fissures toward the ears, hypertelorism, long eyelashes, high arched eyebrows, prominent nose, and malpositioned ears with dysplastic helices. In addition, characteristic skeletal findings are broad short terminal phalanges of the thumbs and halluces, and postnatal growth retardation with head circumference below the fiftieth percentile. Dermatologic features include capillary malformation in approximately 50 percent of patients and higher incidence of keloid formation3 and pilomatricomas4.There may be systemic involvement of multiple organ systems. Of children with RSTS, 24-38 percent have cardiac abnormalities including atrial and ventricularseptal defects, patent ductus arteriosus, coarctation of the aorta, pulmonic stenosis,and bicuspid aortic valve abnormalities5. Feeding difficulties and gastroesophageal reflux disease requires aggressive treatment in young children to prevent nutritional and growth deficits. Cryptorchidism affects 78-100 percent of the male infants6. There is an increased incidence of benign and malignant tumors as well as leukemia and lymphoma3,7. Other organ systems that may be involved include auditory, ophthalmic, endocrine, neurologic, respiratory6 and renal systems.
Rubinsten-Taybi Syndrome is associated with a mutation in the CREB-binding protein gene (CREBBP) located on chromosome 16p 13.3. CREBBP which is essential to normal development. It has been identified as a nuclear protein that participates as a coactivator in cyclic AMP regulated gene expression. The precise relationship between microdeletion in the CREBBP and the phenotype of Rubinstein-Taybi is yet to be elucidated 8,9.
In this report, we introduce a case with features of rubinsten -taybi syndrome with CTEV.
CASE REPORT: A 6-month-old male child presented with CTEV patient came for further management. On examination patient had a distinctive facial appearance with hypertelorism, a broad nasal bridge, a beaked nose,micrognathia, microcephaly, smile grimacing, a narrow and high arched palate and long eye lashes. On examination, short stature, broad thumbs, large toes, and CTEV.
Motor and language developmental delay was noted.
Echo and USG abdomen were done which were normal. Blood investigation done which were within normal limits.
Discussion: Rubinstein-Taybi Syndrome is a rare congenital anomaly. It appears that there is very little documented evidence regarding this syndrome in pediatric literature 9. The cause of RSTS is still unclear, but it is associated with a microdeletion at 16p 13.3 region in the CREB binding protein gene (CREBBP) in some patient, suggesting that deletion is the most probable cause of the syndrome10. CREBBP is a transcription coactivator and functions as a potent histone acetyltransferase, both of which are essential to normal development8. In animal models, the mice with truncated Crb proteindemonstrate clinical features of RSTS observed in humans including growth retardation, retarded osseous maturation, hypoplastic maxilla with a narrow palate, and cardiac and skeletal abnormalities11. More recently, the breakpoint of two distinct reciprocal translocations occurring in patients with the diagnosis of RSTS has been located in the same band 16p 13.3. However, this anomaly cannot be identified in all patients. Clinically, the difference between patients with or without deletion is minimal except for microcephaly. Band 16p 13.3 seems to be an important locus for mental retardation in patients with correct diagnosis of RSTS 6,12,13,14.
Our patient, a known case of CTEV, was identified with characteristic features of the RSTS as explained above. In conclusion, RSTS is a rare genetic condition that affects body shape, extremities, and many organs/systems of the body, particularly cardiac, respiratory, nervous, and uro-genital system. This case report can help pediatricians to become more familiar with this syndrome.
Author’s contribution – Jayant Vagha-guidance, concept ,Ashish Verma- data collection, Abhishek Kalwani-literature search, Ramnath Reddy- manuscript,Anjali Kher -concept,strategy ,
Conflict of interest – Nil
Funding source and its role in the study – No

References:
1. Rubinstein JH, Taybi H. Broad thumbs and toes andfacial abnormalities. Am J Dis Child 1963; 105: 588.
2. Spitz JL. Genodermatosis: a full-color clinical guide to genetic skin disorders. Lippincott, New York: Williams & Wilkins; 1996.P.308-9.
3. Siraganian PA, Rubinstein JH, Miller RW. Keloids and neoplasms in the Rubinstein-Taybi syndrome. Med Pediatr Oncol 1989; 17: 485.
4. Dalal AB, Phadke SR. Morphometric analysis of face in dysmorphology. Comput Methods Programs Biomed 2007 Feb;85(2):165-72.
5. Stevens C, Bhakta M. Cardiac abnormalities in the Rubinstein-Taybi syndrome. Am J Med Genet 1995; 59:346.
6. Wiley S, Swayne S, Rubinstein JH, Lanphear NE, Stevens CA. Rubinstein-Taybi syndrome medical guidelines. Am J Med Genet 2003; 119A:101.
7. Miller R, Rubinstein JH. Tumors in Rubinstein- Taybi syndrome. Am J Med Genet 1995; 56: 112.
8. Roelfsema JH, White SJ, Ariyurek Y, Bartholdi D, Niedrist D, Papadia F, et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet 2005; 76:572-580.
9. Taren Cardona BS, Al Kline DPM. Rubinstein- Taybi Syndrome: A case report. The Foot & Ankle Journal 2008;1(7):3.
10. Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RCM, Masuno M, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional coactivator CBP. Nature 1995; 376: 348-351; PubMed ID: 7630403.
11. Oike Y, Hata A, Mamiya T, Kaname T, Noda Y, Suzuki M, et al. Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: implications for a dominant-negative mechanism. Hum Molec Genet 1999; 8:387-396.
12. Stef M, Simon D, Mardirossian B, Delrue MA, Burgelin I, Hubert C, et al. Spectrum of CREBBP gene dosage anomalies in Rubinstein-Taybi syndrome patients. Eur J Hum Genet 2007; Aug; 15(8):843-7.
13. Petrij F, Dauwerse H, Blough R,Giles R, J van der Smagt J, Wallerstein R. Diagnostic analysis of theRubinstein-Taybi syndrome: five cosmids should be used for microdeletion and low number of protein truncating mutation. J Med Genet 2000; 37:168.
14. Masuno M, Imaizumi K, Ishii T, Kuroki Y, Baba N, Tanaka Y. Pilomatrixomas in Rubinstein-Taybi syndrome. Am J Med Genet 1998; 77: 81.
Issue: October-December 2018 [Volume 7.4]